Next: Automatic editing Up: Methods and Algorithms Previous: Systematic match inspection Contents Index
Reinert et al. (2000) conducted experiments on iterative methods for faster
sum-of-pairs with a divide-and-conquer alignment approach (using a
![]() algorithm) and quasi-natural gap costs for multiple sequence
alignments. Schlosshauer and Ohlsson (2002) devised an algorithm based on fuzzy
recast of dynamic programming algorithms in terms of field annealing to score
the reliability of sequence alignments. These methods also show varying
runtimes ranging from a few seconds to hours for a relatively low number of
sequences, far lower that the realistic number of sequences encountered in
real world assembly projects.
algorithm) and quasi-natural gap costs for multiple sequence
alignments. Schlosshauer and Ohlsson (2002) devised an algorithm based on fuzzy
recast of dynamic programming algorithms in terms of field annealing to score
the reliability of sequence alignments. These methods also show varying
runtimes ranging from a few seconds to hours for a relatively low number of
sequences, far lower that the realistic number of sequences encountered in
real world assembly projects.
Due to these problems, it was decided to use iterative pairwise sequence alignment and devise new methods for searching overlap candidates. A key concept is empowering contigs to accept or reject reads presented to them during the contig building. The algorithm consists mainly of two objects which interact with each other: a pathfinder module and a contig building module.
Hence, one has to make sure to start at the position in the contig where there are as many reads as possible with almost no errors. The pathfinder will thus - in the beginning - search for a node in the weighted graph having a maximum number of highly weighted edges to neighbours. The idea behind this behaviour is to take the read with the longest and qualitatively best overlaps with as many other reads as possible and use this one to ensure a good starting point: the 'anchor' for this contig.
A necessary constraint proved to be that the weight of the edges selected must not fall below a certain threshold as it is not infrequent to have a considerable number of relatively short reads with repetitive element contained in many projects. This introduces a sort of 'gravity well' in the assembly graph and misleads the algorithm to take a read with dangerous repetitive characteristics as anchor, which is generally not the intended behaviour.
The next step consists of determining the next read to add to the now existing contig by analysing nodes (reads) from the contig in the graph and determine which edge (overlap) with a node - not belonging to the contig - provides the most profitable overlap to augment the information contained within the contig.
There are theoretically two distinct approaches to perform this operation:
Extending the contig length first implies the quality of the reads to be reliably high anytime and anywhere. This cannot be guaranteed for sequences extracted by electrophoresis and subsequent base calling as base probability values do not exist for every base calling method. The backbone strategy can also be easily affected by repetitive short and long subsequences in the target DNA if additional assembly support information is not available. On the other hand, once the backbone of the assembly has been constructed, adding more reads to reduce base calling errors step by step is trivial.
Increasing the coverage of a contig first - by adding reads containing mostly sequences already known - ensures that the base qualities of the consensus raise rapidly: more reads that cover a consensus position make consensus errors introduced by bad signal quality and subsequent base calling errors increasingly improbable. Reads added afterwards have thus an increasing probability to align against a right consensus sequence. The disadvantage of this method appears when parts of a contig are only covered by low to medium quality reads. The increased coverage results in a growing number of discrepancies between read bases, leading to incorrect an consensus and sometimes stopping the assembler from adding reads to a contig where there is theoretically a possibility to continue.
![\includegraphics[width=\textwidth]{figures/greedypathgraph}](img133.png) |
The assembly graphs build in the previous step have the unpleasant characteristic to be quite large even for small assemblies. In case of multiple - almost identical - repeats, the number of edges present in the graph explodes. Figure 30 shows a part of a much bigger graph used as example in this section.
![\includegraphics[width=\textwidth]{figures/greedypathass}](img134.png)
|
By performing an in-depth analysis - with a cutoff value of normally 4 or 5 recursions - of the weights to neighbouring reads contained in the assembly graph, the n, m recursive look-ahead strategy can substantially reduce the number of misalignments as chimeric reads get only a very small chance of getting aligned. At the same time, reads containing repetitive elements will not get aligned without 'approval' from reads already in the contig and reads to be aligned afterwards.
To elude these problems known in literature, an alignment has been defined to be optimal when the reads forming it have as few unexplained errors as possible but still form the shortest possible alignment.
For this purpose the contig object has been provided with functions that analyse the impact of every newly added read (at a given position) on the existing consensus. The assumption is now that - as the assembly presumably started with the best overlapping reads available - the bases in the consensus will be right at almost every position. Should the newly added read integrate nicely into the consensus, providing increased coverage for existing consensus bases and perhaps extending it a bit, then the contig object will accept this read as part of the consensus.
In some cases the assembly graph will contain chance overlaps between otherwise unrelated reads. Should such an overlap be good enough to be chosen by the pathfinder algorithm as next alignment candidate, the read will most probably differ in too many places from the actual consensus (thus differing in many aspects from reads that have been introduced to the consensus before). The contig will then reject the read from its consensus and tell the pathfinder object to search for alternative paths through the weighted overlap graph. The pathfinder object will eventually try to add the same read to the same contig but at a different position or - skipping it - try other reads.
Once the pathfinder has no possibilities left to add unused reads to the actual contig, it will again search for a new anchor point and use this as starting point for a new contig. This loop continues until all the reads have been put into contigs or - if some reads could not be assembled anywhere - form single-read contigs.26
The simplest behaviour of a contig could be to simply accept every new read that is being presented as part of the consensus without further checks. In this case the assembler would thus rely only on the weighted graph, the method with which the weighted edges were calculated and the algorithm which traverses this graph. Figure 33 shows this exemplarily for the reads from the example set up above. Although there are four columns having discrepancies, the assembly looks quite reasonable.
![\includegraphics[width=\textwidth]{figures/wrongass}](img136.png)
|
If a newly inserted read shows too many discrepancies to the actual consensus sequence, the contig will invariably reject this read and the edge representing the corresponding overlap will be removed from the assembly graph. The main reasons for overlaps of this type being present in the assembly graph include spurious matches that slipped through Smith-Waterman, repetitive sequence and chimeric reads.
In case of repetitive sequences, each discrepancy between a newly inserted read and the actual consensus is treated as single-base error region, i.e. the explanation of the possible base-call error to be analysed has to be searched in the immediate vicinity of the base. The question the simple signal analysis (see the following section) has to answer is if whether or not it is possible - that is, if there is enough evidence in the trace data - to substitute the offending base of the new read against the base calculated in the consensus. If signal analysis reveals that it is possible to substitute bases, the error is treated as 'explainable' error and it is treated as 'unexplainable' if not.
If the percentage of 'unexplainable' errors in the repetitive region surpasses a certain threshold (default value is 1%), then the newly inserted read will be rejected from the consensus and its node put back into the assembly graph as shown in figure 34.
![\includegraphics[width=\textwidth]{figures/wrongassalu}](img137.png)
|
Two different computational methods have been established in the past years to adjudicate om conflicting bases in reads. The probably most common method is the usage of quality values first introduced by Staden (1989) and further developed by Bonfield et al. (1995a). The concept was then refined to probability values by Ewing and Green (1998). Base probability values are experimentally gained, position-specific probabilities of error for each base-call in a read as a function of certain parameters computed from trace data. Probability values are of high value for finding bases with weak quality, but these values are mostly only available for the called bases, not for uncalled ones. For an assembler, it is a big drawback not to have the capability to search for alternative base-calls that might have been possible at the same position in the trace.
Another suggestion on incorporating electrophoresis data into the assembly process promoted the idea of capturing intensity and characteristic trace shape information and provide these as additional data to the assembly algorithm (Allex et al. (1997,1996)). Although methods for storing alternative base calls and other data have been suggested (Walther et al. (2001)), the approach from Ewing and Green (1998) produces results that are simple to handle and apparently 'good enough' so that it has imposed itself as standard over the years.
Complex shape approaches were discarded early in development of the algorithms for the assembler as essentially all the contig - and with it the consensus computing algorithm of a contig - needs to know is if the signal analysis reveals enough evidence for resolving a conflict between reads by changing the bases incriminated. This question is a simple working hypothesis which is a subset of the hypotheses generated by the automatic editor devised by Pfisterer and Wetter (1999).
It is possible to call the automatic editor in analysis mode to have this specific questions analysed. The conditio sine qua non for the assembly algorithm is that the signal analysis does not try to find support for a change of the possibly faulty base with ``brute force''. This would lead into the doctrine to use high quality sequence to adjust presumably low quality sequence, which is a complete contradiction of the working principle of using high quality data to explain errors in low quality sequences and correct them only if the low quality data supports the new theory.
In fact, the analysis is executed with the uttermost cautiousness and the signal analyser will support a base change only if hard evidence for this hypothesis can be found in the trace signals. Otherwise the assembler treats signal analysis as a black box decision formed by an expert system, only called during the assembly algorithm when conflicts arise. But this expert system provides nevertheless additional and more reliable information than the information contained in quality values extracted from the signal of one read only (compare to Durbin and Dear (1998)): there is a reasonable suspicion deduced from other aligned reads on the place and the type of error where the base caller could have made a mistake by analysing only a single read.
Figure 34 shows this information - along with a new assembly that took place - using the knowledge that repetitive elements must be checked much more strictly. In this new assembly, read 5 and read 4 were rejected from the contig as the pathfinder tried to enter them, because they induced errors into the already existing contig formed by the reads 2-1-3 (when adding read 5) and 2-1-3-6 (when adding read 4).
![\includegraphics[width=\textwidth]{figures/rightass}](img138.png)
|
The contig object failed to find a valid explanation to this problem when calling the signal analysis function - provided by the automatic editor - that tried to resolve discrepancies by investigating probable alternatives at the fault site. Figure 35 shows the result of the assembly that began in figure 34. There is not enough evidence found in the conflicting reads to allow a base change to resolve the discrepancies arising. There is also the additional knowledge that these non-resolvable errors occur in an area tagged as 'ALU-repeat', which leads to a highly sensitive assessment of the errors. Consequently, the reads 4 and 5 - containing repetitive sequence which the first contig object could not make match to its consensus - form a second contig as shown in the example.
The first constraint that can be deduced from this is the fact that - whenever the two reads belonging to a template can be inserted into the same contig - both reads have to be on opposite strands. As the approximate template size is also known, the second - equally important constraint - specifies a size range for both reads belonging to a template to be placed in the contig.
Large projects27 are often sequenced using subclone libraries of different sizes. This can be done purposely to help building assembly scaffolds solely from overlap information and size constraints (Gene Myers' keynote on the German Conference on Bioinformatics (GCB) 99: ''A Whole Genome Assembler for Drosophila''). It can also be a direct implication of the re-sequencing of certain parts of a project.28 The insert size of a template is therefore a template specific information that must be read from experiment files as additional assembly information and cannot be given as general parameter.
If the read is the first of a template to be inserted, the contig will accept it without checking template constraints. In case the newly added read is the second, the contig will check the constraints discussed above and reject the read if either one is not fulfilled.
| = | 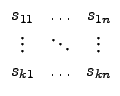 |
| = |
Two more vectors can be defined by recording the error probability for each
base and the direction of each sequence in the alignment:
| = |
| = |
Should discrepancies in the called bases occur in this column, the probability for an error will be computed for each base character group separately. Two main points must be considered when setting up a formula to compute this probability:
As a first step, the probability values can be grouped together by base and direction of the sequence in the alignment:
| c |
| c |
E.g., all bases with an A from sequences in forward direction are taken together in the Pi(A)+ group probability, bases with an A from sequences in reverse complement direction are taken in the Pi(A)- group probability.
Each of the Pi(c) vectors is now sorted by value from low to high. The results are sorted vectors PS of error probabilities, for each base and each direction one.
![\includegraphics[width=\textwidth]{figures/calcgroupqual}](img158.png) |
For the next step, the vectors are pruned to contain a maximum of n lowest probability entries. The motivation for this step is loosely modeled after the entropy calculation theorem of the information theory. Grossly oversimplified, this theorem stipulates that the more information snippets sustaining one single fact are present, the less a single snippet of information itself is worth. Once the probability vector was sorted by value with the lowest values first, taking only the first n lowest values discards only those error probabilities that would not add much to the information itself.
In the next step, the inverse of the probability values are summed up, starting with the highest inverse and modifying the values with a decreasing scalar:
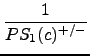 +
+ 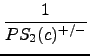 + ... +
+ ... + 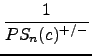
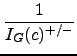
As last step, the total group error probability is computed by multiplying both directed group probabilities:
Once such a group quality has been computed for every base character of a column, the character with the highest group quality wins and is taken as consensus base for this column. When using IUPAC consensus characters, base characters with a certain minimum group quality - e.g. an error probability of less than 0.001 which equals a quality value of 30 and more - are taken to form the IUPAC consensus base.
![[*]](crossref.png)
![\includegraphics[width=8cm]{figures/pathgraph2}](img135.png)